

Research Highlights:
O(N) molecular dynamics for electrochemical systems
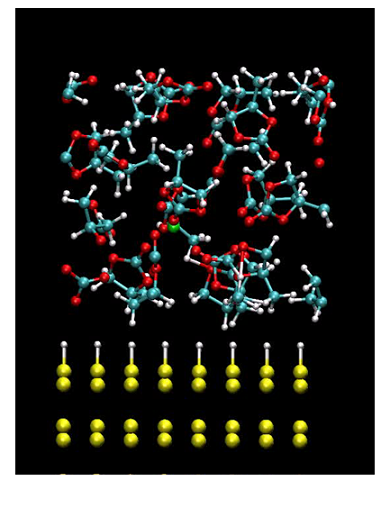
electrolyte of propylene carbonate molecules
Interfaces under an external bias voltage play an important role in a wide variety of chemical reactions such as that in lithium ion batteries. To address such a realistic problem we have developed a method for large-scale first-principles molecular dynamics (MD) simulations on electrochemical systems by combining the effective screening medium (ESM) method [1] with O(N) density functional theory (DFT) [2]. This implementation has been significantly simplified by the introduction of neutral atom potentials, which minimizes the modifications to existing DFT code. In order to demonstrate ability of this implementation, it has been applied to an electrochemical system consisting of a H-Si(111) electrode, which is a candidate anode for high-capacity Li-ion secondary batteries, and a propylene carbonate (PC) solvent to simulate how PC molecules in the vicinity of the electrode surface respond to an imposed electric field. The large-scale MD simulation clearly demonstrates that the combination of the ESM and O(N) DFT methods provides a useful tool for first-principles investigation of complicated electrochemical systems such as high-capacity batteries. The research was conducted in collaboration with NISSAN Research Center, AIST, and Tohoku University.
- "First-principles calculations of charged surfaces and interfaces: A plane-wave nonrepeated slab approach" M. Otani and O. Sugino, Phys. Rev. B 73, 115407 (2006).
- "Large-scale first-principles molecular dynamics for electrochemical systems with O(N) methods", T. Ohwaki, M. Otani, T. Ikeshoji, and T. Ozaki, J. Chem. Phys. 134, 136101 (2012).