

Research Highlights:
電気化学系に対するO(N)第一原理分子動力学計算
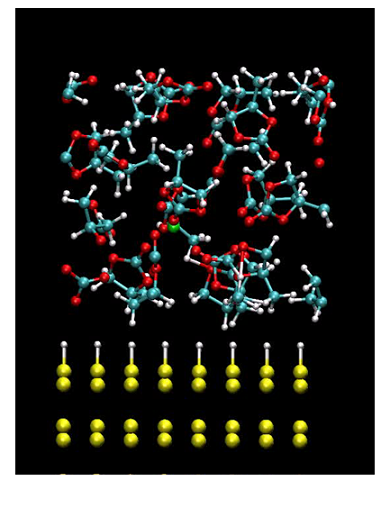
界面構造
電気化学系の電極界面近傍では、実際にはどのような化学反応が進行しているので しょうか。新しい高容量電池の開発を行うためには反応の素過程を詳細に 調べることが必要となってきます。
我々は第一原理計算手法を発展させることで、その 様な複雑な系を取り扱うことの出来る計算手法の開発に取り組みました。 界面近傍での化学反応はバイアス電圧に駆動されて引き起され、ある種の非平衡 状態となっています。また左図に示される様に、電極と多数の分子を含んだ複雑な 系であることも、特徴の一つです。 この様なバイアス電圧下での複雑な系における 化学反応シミュレーションは従来の第一原理計算手法では取扱いが難しいものでした。
我々は有効遮蔽媒質法 [1] とオーダーN密度汎関数法を組合せることにより、 第一原理からこの問題に取り組むことの出来る新しい計算手法を開発しました [2]。 有効遮蔽媒質法 [1] によって、バイアス電圧を実効的に導入することが可能となり、 またKrylov部分空間法に基づくオーダーN密度汎関数法によって、計算コストが 低減され、大規模で複雑な系の取扱いが可能となりました。 一つの応用例として、我々は本手法を用いて、水素で終端されたSi(111)電極と 電解質である炭酸プロピレン分子からなる界面構造のバイアス電圧依存性を 調べました。その結果、バイアス電圧の増加に伴い、炭酸プロピレン分子 がバイアス電圧を打ち消す方向に界面近傍で配向することを見出しました。 今後、我々は超並列大規模計算を実施することで、本手法を反応素過程の解明に 役立てていきたいと考えています。
本研究は日産自動車、産総研、東北大学との共同研究として実施されました。