

�Z�~�i�[ | �ŐV�̃j���[�X | �ߋ��̃j���[�X | �ߋ��̃Z�~�i�[
-
�y2023�N�x�z
- �u�`�S���F ����� ��
- �J�u�����F 11�� 9��(��) 13:20-17:10
- 11��10��(��) 9:20-16:10
- ���{�`���F�Ζʍu�` ���ڍׂɂ��Ă� ������ �����Q�Ƃ��������B
- �J�u�����F
- 9�� 9��(��)�@��1�� 14:00-15:30�A��2�� 15:45-17:15
- 9��16��(��)�@��3�� 14:00-15:30�A��4�� 15:45-17:15
- 9��22��((��))�@��5�� 14:00-15:30�A��6�� 15:45-17:15
- 9��30��(��)�@��7�� 14:00-15:30�A��8�� 15:45-17:15
- ���{�`���F�uZoom�v�𗘗p�����I�����C���u�` ���ڍׂɂ��Ă� ������ �����Q�Ƃ��������B
- �u�`�S���F �����
- �J�u�����F
- 9�� 3��(��)�@��1�� 14:00-15:30�A��2�� 15:45-17:15
- 9��10��(��)�@��3�� 14:00-15:30�A��4�� 15:45-17:15
- 9��17��(��)�@��5�� 14:00-15:30�A��6�� 15:45-17:15
- 9��24��(��)�@��7�� 14:00-15:30�A��8�� 15:45-17:15
- ���{�`���F�uZoom�v�𗘗p�����I�����C���u�` ���ڍׂɂ��Ă� ������ �����Q�Ƃ��������B
- �u�`�S���F �����
- �J�u�����F
- 9�� 4��(��)�@��1�� 14:00-15:30�A��2�� 15:45-17:15
- 9��11��(��)�@��3�� 14:00-15:30�A��4�� 15:45-17:15
- 9��18��(��)�@��5�� 14:00-15:30�A��6�� 15:45-17:15
- 9��25��(��)�@��7�� 14:00-15:30�A��8�� 15:45-17:15
- ���{�`���F�uZoom�v�𗘗p�����I�����C���u�`
- ����F50�����x
- ��u�\�����ԁF2020�N7��2���i�j- 8��21���i���j ���ڍׂɂ��Ă� ������ �����Q�Ƃ��������B
- 2019/8/23�i���j�m���_�Z�~�i�[�n�i�I�����܂����j
- ��ځF Development of Low-order Scaling DFT Methods
- �u���ҁF �����
- �ꏊ�F �����������{�قU�K �Z�~�i�[���T (A615)
- ���ԁF 16:00-17:00
- 2019/7/10�i���j�m���_�C���t�H�[�}���Z�~�i�[�n�i�I�����܂����j
- ��ځFQuasiparticle and optical properties of potential 2D materials
- �u���ҁF Prof. Hung-Chung Hsueh (Department of Physics, Tamkang University, Taiwan)
- �ꏊ�F �����������{�قU�K �Z�~�i�[���T (A615)
- ���ԁF 16:00-17:00
- 2019/7/9�i�j�m���_�C���t�H�[�}���Z�~�i�[�n�i�I�����܂����j
- ��ځFEmbedding the flat bands of Lieb, kagome, and checkerboard lattices into new structures: Tight-binding models to real materials
- �u���ҁF Dr. Chi-Cheng LEE�i�������j
- �ꏊ�F �����������{�قU�K �Z�~�i�[���T (A615)
- ���ԁF 16:00-17:00
- 2019/6/21�i���j�m�����v�]���{�݃Z�~�i�[�n�i�I�����܂����j
- ��ځF�ԗ��IDFT�v�Z�ɂ��AB2�^2���������̍\���}�b�v
- �u���ҁF ���c ���� (�����v�]���{��)
- �ꏊ�F �����������{�قU�K �Z�~�i�[���T (A615)
- ���ԁF 13:00-14:00
- 2019/6/21�i���j�m���_�C���t�H�[�}���Z�~�i�[�n�i�I�����܂����j
- ��ځFShaping Nanostructures Using Molecules
- �u���ҁF Dr. Shih-Hsuan HUNG (������)
- �ꏊ�F �����������{�قU�K �Z�~�i�[���T (A615)
- ���ԁF 16:00-17:00
- 2018/11/6�i�j [LASOR�Z�~�i�[] [���_�Z�~�i�[]�i�I�����܂����j
- ���: Multistep nonradiative decay pathways of biomolecules
- �u�t: �R�� �] ���i���k��w�����ޗ��������j
- �ꏊ: �����������{�قU�K ��5�Z�~�i�[��
- ����: 16:00-17:00
- 2018/6/1�i���j ���_�Z�~�i�[�i�I�����܂����j
- ���: �g�|���W�J���M�d�ϊ������f�U�C���������ُ�l�����X�g�W���̑�ꌴ���v�Z
- �u�t: �Έ�@�j�V �� / �����w��w�@ ���R�Ȋw������ �����Ȋw��U, ������w����������
- �ꏊ: �����������{�قU�K A615����
- ����: 16:00-17:00
- 2018/2/21�i���j ���_�C���t�H�[�}���Z�~�i�[�i�I�����܂����j
- ���: Photoinduced reaction dynamics of nanocarbons
- �u�t: Dr. Kaoru Yamazaki / Institute for Materials Research, Tohoku Univ.
- �ꏊ: �����������{�قU�K A615����
- ����: 13:30-14:30
- 2017/12/1�i���j ���_�Z�~�i�[�i�I�����܂����j
- ���: Recent Progress in a Calculation Method of Quasiparticle Spectra
- �u�t: ��삩���鎁 / ���l������w��w�@�H�w�����@
- �ꏊ: �����������{�قU�K A615����
- ����: 16:00-17:00
- 2017/8/29�i�j ���_�Z�~�i�[�i�I�����܂����j
- ���: Skyrmion crystal as a promising thermoelectric converter: A prediction from first-principles
- �u�t: Mr. Yo Pierre Mizuta / Graduate School of Natural Science and Technology, Kanazawa University
- �ꏊ: �����������{�قU�K A615����
- ����: 14:00-15:00
- 2017/7/28�i���j���_�C���t�H�[�}���Z�~�i�[�i�I�����܂����j
- ��ځFOrigin of the spin reorientation transitions in antiferromagnetic MnPt-based alloys�i�I�����܂����j
- �u�t�FDr. Po-Hao Chang / Department of Physics and Astronomy, University of Nebraska-Lincoln
- �ꏊ: �����������{�قU�K A615����
- ����: 16:00-17:00
- 2017/5/30�i�j���_�Z�~�i�[�i�I�����܂����j
- ��ځFAbsolute binding energies of core levels in solids from first principles�i�I�����܂����j
- �u�t�F���� �� / ������w����������
- �ꏊ: �����������{�قU�K A615����
- ����: 16:00-17:00
- 2017/2/10�i���j���_�Z�~�i�[�i�I�����܂����j
- ��ځFRare-earth magnet�i�I�����܂����j
- �u�t�F�O�� �� �� / �Y�ƋZ�p����������, �����E�ޗ������@�\
- �ꏊ: �����������{�قU�K A615����
- ����: 16:00-17:00
- 2016/8/26�i���j���_�C���t�H�[�}���Z�~�i�[�i�I�����܂����j
- ��ځF��ꌴ���d�q��Ԍv�Z��p�����V������M�d�ޗ��̕����𖾂ƃ}�e���A���f�U�C��
- �u�t�F�{�c �S�W �� / �k����[�Ȋw�Z�p��w�@��w ��[�Ȋw�Z�p������
- �ꏊ: �����������{�قU�K A615����
- ����: 16:00-17:00
- 2016/6/24�i���j���_�C���t�H�[�}���Z�~�i�[�i�I�����܂����j
- ��ځFLocal physical quantities for spin based on quantum�@electrodynamics
- �u�t�F���c ���� / ������w����������
- �ꏊ: �����������{�قU�K A615����
- ����: 16:00-17:00
- 2016/5/20�i���j���_�Z�~�i�[�i�I�����܂����j
- ��ځF�A�����t�@�X�����_�����ɂ��镁�ՓI�Ȓ����ƕs�K���\���̋L�q�@
- �u�t�F���� ���� �� / �Y�ƋZ�p�����������@�\�ޗ��R���s���e�[�V���i���f�U�C�������Z���^�[
- �ꏊ: �����������{�قU�K A615����
- ����: 16:00-17:00
- 2016/4/26�i�j���_�C���t�H�[�}���Z�~�i�[�i�I�����܂����j
- ��ڇ@�F�W�c�^���ƏW�c�I�ӎv����̃��f��
- �u�t�F���� ���V �� / ������w�H�w�n�����ȕ����H�w��U
- �ꏊ: �����������{�قU�K A612����
- ����: 13:30-14:10
- ��ڇA�FPolyexciton stability in multi-valley semiconductor and optical trap for valley exciton
- �u�t�F���� �m�� �� / ������w���w�n�����ȕ����w��U
- �ꏊ: �����������{�قU�K A612����
- ����: 14:20-15:00
- 2015/11/27�i���j���_�Z�~�i�[�i�I�����܂����j
- ��ځFRepresentation of first-principles band structures in a conceptual Brillouin zone
- �u�t�FChi-Cheng Lee / ������w����������
- �ꏊ: �����������{�قU�K A615����
- ����: 16:00-17:00
- 2015/8/7�i���j��3��v�Z�����Ȋw�Z�~�i�[�i�I�����܂����j
- ��ځFFirst-principles study of large spin-orbit coupling transition-metal compounds: electronic structure and new possibilities
- �u�t�FMyung Joon Han / Assist. Prof. Department of Physics, KAIST
- �ꏊ: CMSI�_�ˋ��_�i�����w�������E�v�Z�Ȋw�����@�\�E5�K501�����j
- ����: 14:00-15:00
- 2015/7/24�i���j���_�Z�~�i�[�i�I�����܂����j
- ��ځFInteratomic potential of iron based on machine learning
- �u�t�F���� �� / ������w����������
- �ꏊ�F�����������{�قU�K A615����
- ���ԁF16:00-17:00
- 2015/5/14�i���j��2��v�Z�����Ȋw�Z�~�i�[�i�I�����܂����j
- ��ځFLarge-scale ab initio simulations based on systematically improvable atomic basis
- �u�t�FProf. Lixin He / University of Science and Technology of China
- �ꏊ: CMSI�_�ˋ��_�i�����w�������E�v�Z�Ȋw�����@�\�E5�K501�����j
- ����: 11:00-12:20
- 2015/1/28�i���j��2��v�Z�����Ȋw�Z�~�i�[�i�I�����܂����j
- ��ځF�S�d�q�t���|�e���V�������`���⋭���ʔg�@�ɂ��J�ڋ������������̗��_�I�\��
- �u�t�F�����_�� �� / �O�d��w��w�@�H�w������ �y����
- �ꏊ: CMSI�_�ˋ��_�i�����w�������E�v�Z�Ȋw�����@�\�E5�K501�����j
- ����: 13:30-14:30
- 2014/10/31�i���j���_�Z�~�i�[�i�I�����܂����j
- ��ځFTheory Seminar:Theoretical and Experimental Exploration of Two-Dimensional Silicon Structures
- �u�t�F���� �� / ������w����������
- �ꏊ�F�����������{�قU�K A615����
- ���ԁF16:00-17:00
- The 4th OpenMX developer's meeting
�y2022�N�x�z
- ���I�����C���u�`�� 9���J�u/�S8��u�}�e���A���Y�E�C���t�H�}�e�B�N�X�̊�b�Ɖ��p�v
�y2021�N�x�z
- ���I�����C���u�`�� 9���J�u/�S8��u��ꌴ���d�q��Ԍv�Z�̊�b�Ɖ��p�v
�y2020�N�x�z
- ���I�����C���u�`�� 9���J�u/�S8��u��ꌴ���d�q��Ԍv�Z�̊�b�Ɖ��p�v
To further extend the applicability of first-principles electronic structure calculations based on density functional theory (DFT) to large-scale systems containing more than ten thousands of atoms, here we present development of low-order scaling DFT methods: one is a numerically exact one, the other is approximate O(N) methods. Though the conventional DFT calculations based on semi-local functionals scale as the third power of number of atoms, it will be shown that the computational complexity of DFT calculations can be reduced to a low-order scaling in a numerically exact sense [1,2]. We further discuss an efficient O(N) divide-conquer (DC) method based on localized natural orbitals (LNOs) for large-scale DFT calculations of gapped and metallic systems [3], where the LNOs are noniteratively calculated by a low-rank approximation via a local eigendecomposition of a projection operator for the occupied space. In addition to the low-order scaling methods, efficient parallelization methods for massively parallel computers will be presented for atom decompositions [4] and fast Fourier transforms [5,6].
References:[1] T. Ozaki, Phys. Rev. B 82, 075131 (2010).
[2] T. Ozaki, Phys. Rev. B 75, 035123 (2007).
[3] T. Ozaki, M. Fukuda, and G. Jiang, Phys. Rev. B 98, 245137 (2018).
[4] T.V.T. Duy and T. Ozaki, Comput. Phys. Commun. 185, 777 (2014).
[5] T.V.T. Duy and T. Ozaki, Comput. Phys. Commun. 185, 153 (2014).
[6] T.V.T. Duy and T. Ozaki, J. Supercomput. 72, 391 (2016).
�������̃Z�~�i�[�y�[�W �����Q�Ƃ��������B
�y2019�N�x�z
Quasi-2D atomically thin materials display a number of unique properties not found in their bulk counterparts, such as large self-energy and excitonic effects due to the quantum confinement and reduced screening with layer number close to the 2D limit. These atomically thin layer structures demonstrate rich physics and pave the way for emerging fields, such as excitonics and valleytronics, with great potential for applications in next-generation devices. To probe the dimensionality effects, we use ab initio GW+BSE methods based on many-body perturbation theory (MBPT) to explain and predict the quasiparticle and optical properties of potential quasi-2D semiconductors: monolayer group VI monochalcogenides (Ge,Sn/S,Se) and transition metal dichalcogenides (MoSe2 and Janus MoSSe). Significant exciton binding energy, layer-controlled bandgap, anisotropic optical response [1], and possible valley polarization [2] serve as a convenient and efficient method for engineering the excited-state properties of quasi-2D systems.
References:
[1] Hung-Chung Hsueh, Jia-Xuan Li, and Ching-Hwa Ho, Adv. Optical Mater. 6, 1701194 (2018).
[2] Ang-Yu Lu, et.al, Nature Nanotechnology 12, 744 (2017).
�������̃Z�~�i�[�y�[�W �����Q�Ƃ��������B
"Embedding the flat bands of Lieb, kagome, and checkerboard lattices into new structures: Tight-binding models to real materials"
The studies of dispersion-less bands revealed in electronic and photonic systems have caught great attention recently. Many exotic quantum phenomena, for example, the high-transition-temperature superconductivity associated with the infinitely large density of states of the flat bands, are proposed. In this talk, I will begin with an introduction to the flat bands using Wannier functions. Then I will introduce three tight-binding models, namely the Lieb, kagome, and checkerboard lattices, by considering only the nearest-neighbor hopping parameters and demonstrate that the recognized flat bands associated with the three lattices can be ideally embedded into new structures, respectively [1]. Finally, I will provide several examples for the appearance of nearly flat bands realized in two-dimensional materials with long-range hopping beyond the simplified tight-binding models based on our first-principles calculations for the systems composed of Ge atoms.
Our study clearly demonstrates that the flat bands given by the well-known lattices, namely the Lieb, kagome, and checkerboard lattices, can be ideally embedded into the new structures that cannot be recognized as the original ones. Therefore, the amount of materials that can give interesting flat-band physics could be much larger.
References:
[1] Chi-Cheng Lee et al., arXiv:1904.07048 (2019).
�������̃Z�~�i�[�y�[�W �����Q�Ƃ��������B
�w�ԗ��IDFT�v�Z�ɂ��AB2�^2���������̍\���}�b�v�x
���̏\���N��2���������͂��̑��l�ȕ����ƕ��L�����p�\�����瑽���̒��ڂ��W�߂Ă���B���ɋߔN�̔����E�����Z�p�̔��W�ɂ��A1T(trigonal monolayer) �� 1H (hexagonal monolayer)�Ƃ����������I�ȍ\��������AB2�^�̑J�ڋ����_�C�J���R�Q�i�C�h(e.g. MoS2) ��MXenes (�J�ڋ����J�[�o�C�h/�i�C�g���C�h) �ȂǂɊւ��錤������������Ă���B������AB2�^�P�w���������ɕ���Ă��邪[1]�AAB2�^�̉����������ł����Ă����f�̑g�ݍ��킹�͖c��ł��邽�߁A�܂��܂��v�Z�����Ȋw�̗��ꂩ�疢�m�̂Q����������T������]�n���\���ɂ���ƍl������B�����Ŗ{�����ł�OpenMX[2]��p�����ԗ��IDFT�v�Z�ɂ��AB2�^2���������̍\���}�b�v���쐬����[3,4]�B��̓I�ɂ́A��K�X����^�m�C�h��A�N�`�m�C�h��������62���f�̑g�ݍ��킹����Ȃ�3844������(=62���f�~62���f)�ɑ��ď����\���Ƃ���1T�E1H�Eplanar��3��ނ�p�ӂ��A2�~2�̃X�[�p�[�Z���ɑ��č\���œK�����s���A����ꂽ�\������ԌQ�ŕ��ނ��邱�Ƃɂ��\���}�b�v���쐬�����B�܂��A���̍\���}�b�v��p���Ď����\�̑��̑g�ݍ��킹���Ƃ�1T/1H�\�������������̐����܂Ƃ߂邱�Ƃɂ��A1T/1H�\���ɂȂ�₷�����̑g�ݍ��킹�𖾂炩�ɂ����B����ɁA���q�T�C�Y�̃o�C�i���L���f�q�Ƃ��ċ@�\����ƍl������u�������\���v��A���̑������I�Ȗ��m��AB2�^2�����������\���œK���ɂ�蓾��ꂽ�B
���̂悤�ȍ\���}�b�v�́A���m��2���������̍\���T���ɐV�������_��^���A����Ȃ�����I�ȍ\���T���𑣐i����ƂƂ��ɐV�K�����̒T���ɂ����ɗL�p�ł���ƍl������B
References:
[1] J. Zhou et al., Nature 556, 355 (2018).
[2] OpenMX
[3] M. Fukuda, J. Zhang, Y.-T. Lee, T. Ozaki, arXiv:1904.06047 [cond-mat.mtrl-sci]
[4] Our structure map and database on interactive website
"Shaping Nanostructures Using Molecules"
Metallic nanoparticles are widely used for technological applications in catalysis, data storage and solar energy. However, the performance of nanoparticles is usually determined by the shape of nanoparticles. Therefore, understanding the morphology and composition of the metallic nanoparticles changed by environment is important. In this presentation, we will discuss the external factors, such as adsorbed molecules and substrate material, on nanoparticle using density functional theory (DFT) calculations. First, we present the morphology changing of L10 ordered FePt epitaxial growth on Mg(1-x)TixO substrates [1]. Second, we demonstrate the investigation on Ti nanoparticles oxidation, strain and oxygen penetration [2]. Next, we investigate the atomic arrangement of TiPt nanoparticles under different oxygen adsorption [3]. Finally, we study the strong metal-support interaction (SMSI) between Au nanoparticles and ZnO substrates and partly explain the enhanced catalytic reaction (CO oxidation) by the ZnO encapsulation. The investigations show computational calculations can be used to model modification of nanoparticles by adsorbed molecules or supports, and study the properties changing, such as morphology, energy barrier, atomic arrangement and catalytic performance. In summary, the study demonstrates the functional characteristics of nanoparticles highly depend on their nanostructures.
References:
[1] S-H. Hung and K. P. McKenna, Phys. Rev. Materials 1, 024405 (2017).
[2] S-H. Hung and K. P. McKenna, J. Phys. Chem. C 122, 3107 (2018).
[3] S. Gholhaki, S-H. Hung, D. J. H. Cant, C. E. Blackmore, A. G. Shard, Q. Guo, P. McKenna and R. E. Palmer, RSC Adv. 8, 27276 (2018).
�������̃Z�~�i�[�y�[�W �����Q�Ƃ��������B
�y2018�N�x�z
| ����: | (July 2nd-6th) The Summer School on DFT: Theories and Practical Aspects |
| (July 9th and 10th) The 3rd OpenMX developer's meeting (July 11th and 12th) Advanced Lecture Series |
|
| �ꏊ: | �����������{�قU�K |
Nonradiative decay (NRD) processes such as internal conversion (IC) and intersystem crossing (ISC) govern photo-functionality of living systems such as the photostability of the DNA, phototaxis, visual perception etc. Understanding this complex NRD pathways of biomolecules should help us develop bio-inspired photo-functional materials. In this talk, we will share our recent theoretical works on the multistep NRD pathways of biomolecules in gas phase with the aids of pump-probe experiments:
(1) Structure dependent photoisomerization routes of cinnamate based sunscreens1,2
We found that the structural isomers of cinnamate based sunscreens undergo trans �� cis photo-isomerization under UV irradiation but differ in the isomerization pathway (ISC or IC) 1,2. These results suggest that controlling the substitution position is essential to design the cinnamate based photo-functional materials.
(2) Ultrafast nonadiabatic cascade of XUV excited caffeine molecule3
We also found the XUV excited caffeine molecule undergoes a nonadiabatic cascade via ~100 highly correlated mono-cationic states. These results show that both nonadiabatic coupling and electron correlation are the keys for ultrafast reaction dynamics in the highly-correlated electronic excited states.
References:
[1] K. Yamazaki, S. Maeda*, M. Ehara*, T. Ebata* et al., J. Phys. Chem. Lett. 7, 4001 (2016).
[2] S. Kinoshita, K. Yamazaki*, M. Ehara*, T. Ebata* et al., Phys. Chem. Chem. Phys. 20, 17583 (2018).
[3] A. Marciniak, K. Yamazaki*, F. Lepine* et al., J. Phys. Chem. Lett., submitted (2018).
*Corresponding authors
�w�g�|���W�J���M�d�ϊ������f�U�C���������ُ�l�����X�g�W���̑�ꌴ���v�Z�x
�V���ȕ����̋N���Ƃ��āC�d�q��Ԃ̃g�|���W�[�����ڂ𗁂тĂ���B�����̑�ꌴ���d�q��Ԍv�Z�v���O�����ɏ��߂ē������ꂽBloch�g�����̃g�|���W�[�Ɋ֘A�����v�Z��@�́C1993�N�ɒ�Ă��ꂽ�d�C���ɂ��v�Z����King-Smith��Vanderbilt�ɂ��x���[�ʑ��̕��@[1]�ł���B��X�͂���܂ŁC�x���[�ʑ��̕��@�C������g�������x���[�ȗ��C�g�|���W�J���s�ϗʁiChern��, Z2���j�̌v�Z��@�̑�ꌴ���d�q��Ԍv�Z�v���O�����ւ̎����Ƃ��̉��p[2-5]�Ɏ��g��ł����B�{�u���ł́C�����̔M�d�ϊ����ۂւ̉��p��ɂ��ďЉ��B
��X�́C�M�d�ϊ��ޗ��̍���������͍�����V���ȕ������Ƃ��āC�x���[�ȗ��R���ُ̈�z�[���W���̃t�F���~���ʈˑ������d�v�ƂȂ�C�ُ�l�����X�g�W���̑傫�Ȍn�̕����f�U�C�����߂����Ă���B���̎��g�݂Ƃ��āC�X�J�[�~�I���������f��[4], �_���������X�J�[�~�I������[6], �n�[�t�z�C�X���[��������[7]�ɂ��Ẳ�X�̍ŋ߂̌������Љ�C�x���[�ȗ����U�N���鋐��ȔM�d�ϊ����ʂ���������, �g�|���W�J���M�d�ϊ������f�U�C���w�j�̍\�z�ɂ��Ă̓W�]���q�ׂ�B
[1] R.D. King-Smith and D. Vanderbilt, Phys. Rev. B, 47, 1651(1993).
[2] F. Ishii and T. Oguchi, J. Phys. Soc. Jpn., 71, 336 (2002).
[3] F. Ishii, N. Nagaosa, Y. Tokura, and K. Terakura, Phys. Rev. B 73, 212105 (2006).
[4] Y. P. Mizuta and F. Ishii, Sci. Rep., 6, 28076(2016).
[5] H. Sawahata, N. Yamaguchi, H. Kotaka, and F. Ishii, Jpn. J. App. Phys., 57, 030309 (2018).
[6] Y.P. Mizuta, H. Sawahata, and F. Ishii, arXiv:1803.08148.
[7] S. Minami, F. Ishii, Y.P. Mizuta, and M. Saito, arXiv:1804.00297.
�y2017�N�x�z
Nanocarbons such as fullerene, carbon nanotube, and graphene are the fundamental materials for carbon-based nanotechnologies. Their optical and electronic properties heavily depend on their size and shape. In order to realize single-molecule scale structural engineering of nanocarbons using laser irradiation, we quantum-chemically investigated the mechanism of the photoinduced reaction dynamics of nanocarbons both in energy and time domains.
We first investigated the reaction paths of Stone?Wales rearrangement (SWR), i.e., ��/2 rotation of two carbon atoms with respect to the midpoint of the bond, in graphene and carbon nanotube at the MS-CASPT2//SA-CASSCF level of multi-reference molecular orbital theory [1]. We found that the vibronic (electron-phonon) coupling play a crucial role to reduce the effective reaction barriers of the photoinduced defect formation of nanographene.
We next investigated that the fragmentation dynamics of the highly charged fullerene cation C60q+ (q = 20-60) produced by the irradiation of x-ray free electron laser pulse using on-the-fly classical trajectory calculations combined with density functional based tight-binding theory. We found that a two-step explosion mechanism governs the fragmentation dynamics [2]: C60q+ firstly ejects singly and multiply charged fast atomic cations Cz+ (z ? 1) to reduce its strong intramolecular Coulomb repulsion on a timescale of 10 fs. Thermal (statistical) evaporations of slow atomic and molecular fragments from the remaining core cluster subsequently occur on a timescale of 100 fs to 1 ps.
I will also briefly discuss our recent results on the real-time imaging of the near-/mid-IR induced coherent vibration of C60, which is considered as the initial step of the photoinduced fragmentation of C60 [3]
References:
[1] K. Yamazaki et al., J. Phys. Chem. A 116, 11441 (2012).
[2] K. Yamazaki et al., J. Chem. Phys. 141, 121105 (2014).
[3] K. Yamazaki et al., to be submitted.
�wRecent Progress in a Calculation Method of Quasiparticle Spectra�x
Photoelectron spectra represent the total energy difference between the N-electron system and the (N�}1)-electron system, which give the basic idea of the quasiparticle (QP) energies.
Very recently, we found that the rigorous formulation involving the QP energies, the QP wave functions, and the QP equation can be applied not only to the N-electron ground state but also to any M-electron excited eigenstate [1]. Focusing on this topic, I will explain our recent achievement of the self-consistent GW�� calculation [2], the GW without Bethe-Salpeter equation method for photoabsorption spectra [3], and the TDGW method for excited state dynamics simulations [4]. These calculations were carried out by using the all-electron mixed basis approach (program name TOMBO), which uses both numerical atomic orbitals and plane waves [5].
[1] K. Ohno, S. Ono, and T. Isobe, J. Chem. Phys. 146, 084108 (2017).
[2] R. Kuwahara, Y. Noguchi, and K. Ohno, Phys. Rev. B 94, 121116(R) (2016).
[3] T. Isobe, R. Kuwahara, and K. Ohno, to be submitted.
[4] T. N. Pham, S. Ono, and K. Ohno, J. Chem. Phys. 144, 144309 (2016).
[5] S. Ono, Y. Noguchi, R. Sahara, Y. Kawazoe, and K. Ohno, Comp. Phys. Comm 189, 20 (2015).
�wSkyrmion crystal as a promising thermoelectric converter: A prediction from first-principles�x
The anomalous Nernst effect (ANE), a heat-to-electric conversion in mutually transverse directions, can be driven by an emergent magnetic field B originating from inhomogeneous magnetic moments in solids. Large ANE has been experimentally confirmed in various ferromagnets, and only very recently, also in an antiferromagnet [1]. Here we theoretically propose that, the Skyrmion crystal (SkX), in which magnetic topological objects Skyrmions are crystallized, is another candidate to host large ANE.
We have found through first-principles calculations on a single s-orbital model using the package OpenMX [2] and Wannier90 [3] that, in a two dimensional SkX phase a large ANE would appear when chemical potential �� is properly tuned (Figure) [4]. This was interpreted as due to its characteristic distribution of Chern numbers among the bands (each Chern number representing quantized flux of B field through each electronic band in momentum space).
Following such an observation in the simplest model of square SkX [4], our subsequent computations on a more realistic oxide film also predict large ANE.
This motivates further studies of ANE in the SkX family, in quest of better thermoelectric materials that exploit this effect.
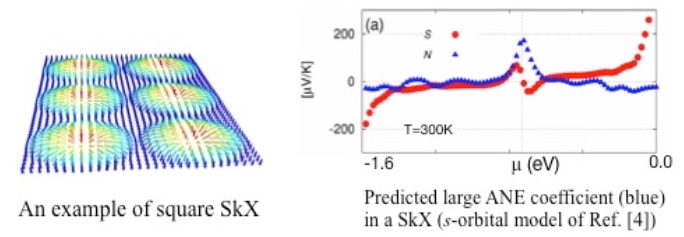
[1] M. Ikhlas et al., Nature Physics (2017) doi:10.1038/nphys4181.
[2] T. Ozaki et al., Open source package for Material eXplorer,�@http://www.openmx-square.org/
[3] A. A. Mostofi et al., http://www.wannier.org.
[4] Y. P. Mizuta and F. Ishii, Scientific Reports 6, 28076 (2016).
�wOrigin of the spin reorientation transitions in antiferromagnetic MnPt-based alloys�x
Antiferromagnetic MnPt exhibits a spin reorientation transition (SRT) as a function of temperature, and offstoichiometric Mn-Pt alloys also display SRTs as a function of concentration. Here we describe the origin of these SRTs using first-principles calculations based on the coherent potential approximation, treating chemical and thermally-induced spin disorder on equal footing. We find that the experimentally observed SRTs are related to specific features in the band structure, and we perform a detailed analysis of the effects of temperature and concentration on the magnetocrystalline anisotropy.
Reference:
P.-H. Chang, T. Markussen, S. Smidstrup, K. Stokbro, and B.K. Nikolic, Phys. Rev. B 92, 201406(R) (2015).
J.M. Marmolejo-Tejada, K. Dolui, P. Lazic, P.-H. Chang, S. Smidstrup, D. Stradi, K. Stokbro, B.K. Nikolic, arXiv:1701.00462.
�wAbsolute binding energies of core levels in solids from first principles�x
The X-ray photoelectron spectroscopy (XPS) is one of the most important and widely used techniques in studying chemical composition and electronic states in the vicinity of surfaces of materials. In spite of the long history of XPS and its importance in materials science, a general method has not been developed so far to calculate absolute binding energies for both insulators and metals, including multiple splittings due to chemical shift, spin-orbit coupling, and exchange interaction, on equal footing. Here, we propose a general method to calculate absolute binding energies of core levels in metals and insulators, based on a penalty functional and an exact Coulomb cutoff method in a framework of the density functional theory [1]. It is demonstrated that the absolute binding energies of core levels for both metals and insulators are calculated by the proposed method in a mean absolute (relative) error of 0.4 eV (0.16 %) for eight cases compared to experimental values measured with XPS within a generalized gradient approximation to the exchange-correlation functional. Recent applications of the method including silicene [2], borophene, and platinum atoms will also be discussed in comparison with experimental data together with analysis of the initial and final state effects based on an energy decomposition method.
[1] T. Ozaki and C.-C. Lee, Phys. Rev. Lett. 118, 026401 (2017).
[2] C.-C. Lee, J. Yoshinobu, K. Mukai, S. Yoshimoto, H. Ueda, R. Friedlein, A. Fleurence, Y. Yamada-Takamura, and T. Ozaki, Phys. Rev. B 95, 115437 (2017).
�y2016�N�x�z
Rare-earth magnets are mainly composed of 3d transition metals (T) and rare-earth metals (R). The former yield large magnetization, while the latter are a source of strong magnetocrystalline anisotropy (at low temperature). Strong magnet compounds, such as Nd2Fe14B and Sm2Fe17N3, contain a light element (X) as a third element. We will discuss the role of the X element in the magnetism of R-T-X systems. First-principles calculations [1] clarify that the magnetic moment depends sensitively on X as a consequence of orbital hybridization between X-2p and T-3d. Crystal-field coefficients at the R sites are also affected by X. This suggests that magnetocrystalline anisotropy can be controllable by additive elements. We will also present a combined first-principles and classical spin model analysis of magnetocrystalline anisotropy at finite temperature [2,3,4].
[1] Y. Harashima et al., Phys. Rev. B 92, 184426 (2015).
[2] M. Matsumoto et al., J. Appl. Phys. 119, 213901 (2016).
[3] Y. Toga et al., Phys. Rev. B 94, 174433 (2016).
[4] T. Fukazawa et al., arXiv:1612.04478.
�w��ꌴ���d�q��Ԍv�Z��p�����V������M�d�ޗ��̕����𖾂ƃ}�e���A���f�U�C���x
�M�d���d�Ƃ̓[�[�x�b�N���ʂ𗘗p���ĔM�G�l���M�[��d�C�G�l���M�[�ɕϊ�����Z�p�ł���C���x�̒Ⴂ���R�G�l���M�[��p�M��p���Ĕ��d���s���g�G�l���M�[�n�[�x�X�e�B���O�h�̒��ł��d�v�Ȉʒu���߂�Ɗ��҂���Ă���D�M�d���d�f�q�Ƃ��ĉ��p����Ă���Bi2Te3��PbTe�͊��fTe���܂ނ��߁CTe���܂܂Ȃ��M�d�ޗ��̊J�������߂��Ă���D����16���Œn�k�ɖL�x�ɑ��݂��闰��S��Te�̑���f�Ƃ��ėL�]�ł���CS���听���Ƃ����������M�d�ޗ��̌��������͓I�ɍs���Ă���B
��X�͗������ɒ��ڂ��C��ꌴ���d�q��Ԍv�Z�Ǝ����̗��ʂ���J�ڋ����������E���}�i�C�gNiSbS������ȏo�͈��q���������Ƃ𖾂炩�ɂ���[1]�D��ꌴ���d�q��Ԍv�Z�\�t�g�E�F�A�p�b�P�[�WOpenMX[2]�C�����_�E�A�[���_�Ɋ�Â��d�q�A���v�Z�c�[��QTWARE[3]��p�����ڍׂȓd�q�\���̉�͂���NiSbS�̍����o�͈��q�̋N���͉��w�|�e���V�����ʋߖT�̋[�M���b�v�\���ł��邱�Ƃ𖾂炩�ɂ����D�{�u���ł͂����̏ڍׂɂ��Ĕ��\����ƂƂ��ɁC��ꌴ���d�q��Ԍv�Z��p�����V������M�d�ޗ��̃}�e���A���f�U�C���ɂ��Ă��Љ��[4]�B
[1] M. Miyata, T. Ozaki, S. Nishino, and M. Koyano, (submitted to J. J. Appl. Phys.).
[2] T. Ozaki, Phys. Rev. B 67, 155108 (2003).
[3] http://www.rs.tus.ac.jp/takahiro/QTWare.html�D
[4] M. Miyata, T. Ozaki, and M. Koyano, (submitted to J. Electron. Mater.).
�wLocal physical quantities for spin based on quantum�@electrodynamics�x
The local picture of electron spin is studied in the framework of quantum electrodynamics(QED). In the framework of QED, one of the fundamental local physical quantities is the energy-momentum tensor density, which is derived from the general principle of relativity.
Recently, the �gquantum spin vorticity theory�h[1] is proposed as a consequence of the general relativistic symmetry of the energy-momentum tensor.
The quantum spin vorticity theory can give the time evolution equations of the electronic momentum density and the spin angular momentum density as equations which relate local mechanical physical quantities derived from the energy-momentum tensor density.
These local images of an electronic state by the quantum spin vorticity theory can help us to understand spin phenomena in condensed matter and molecular systems from a unified viewpoint [2,3].
[1] A. Tachibana, J. Math. Chem. 50, 669 (2012); Electronic Stress with Spin Vorticity. In Concepts and Methods in Modern Theoretical Chemistry, S. K. Ghosh and P. K. Chattaraj. Eds., CRC Press, Florida (2013), pp. 235-251; J. Comput. Chem. Jpn. 13, 18 (2014); Indian J. Chem. A, 53, 1031 (2014).
[2] M. Fukuda, K. Soga, M. Senami, A. Tachibana, Int. J. Quant. Chem., 116, 920 (2016)
[3] M. Fukuda, K. Ichikawa, M.Senami, A. Tachibana, AIP Advances 6, 025108 (2016).
�w�A�����t�@�X�����_�����ɂ��镁�ՓI�Ȓ����ƕs�K���\���̋L�q�@�x
HfO2�AZrO2, TiO2, In2O3, Ga2O3, Al2O3,Cu 2O, Li2O�Ȃǂ̃A�����t�@�X�����_�����̌��q�\�����I�ɑ������u��d�����_���[�U�\���v�Ƃ����l���������A�ꌩ���G�Ɍ�����A�����t�@�X�����_�����ɂ͒P���ŕ��ՓI�Ȓ��������������݂��邱�Ƃ��������[1,2]�B�����āA���������������������n���I�ɒ��ׂ邽�߂ɕK�v�Ȍ��t�����@���Ă��邱�Ƃ��w�E���A���̖����������邽�߂ɑn�o���������I��@���������[3,4]�B�s�K�����q�z����{���m�C���ʑ̃^�C�����O�ŕ\���ł��邪�A�\���v�f�ł���{���m�C���ʑ̂̔z��p�^�[���𐔗�ŕ\���ł���A�Ή����錴�q�z���\���������ƂɂȂ�B�{��@�́A���ʑ̂��\�����鑽�p�`�̑g�ݍ��킹�Ɋւ���K���𗘗p���āA���ʑ̂𑽖ʑ̃R�[�h���[�h�Ƃ�������ŕ\���B���̍l�������g�����āA�{���m�C���ʑ̃^�C�����O�𑽖ʑ̃R�[�h���[�h�̕��сi���E�̃R�[�h���[�h�Ƃ����j�ŕ\���B
[1] Universal Medium-Range Order of Amorphous Metal Oxides, PRL 111, 15502 (2013).
[2] Dual-Random-Sphere-Packing Structure of Liquid and Amorphous Li2O, Trans. Mat. Res. Soc. Japan, 40, 141-144 (2015).
[3] How to describe disordered structures, Sci. Rep. 6, 23455.
[4] �A�����t�@�X�ޗ��Ȃǂ̕s�K���Ȍ��q�z���\�����鐔���I��@��n�o
http://www.aist.go.jp/aist_j/press_release/pr2016/pr20160411/pr20160411.html
�w�W�c�^���ƏW�c�I�ӎv����̃��f���x
�����̌Q��^���𐔗����f���ɂ���ăV�~�����[�g���鎎�݂́A1980�N��ɂ��̖G�肪�����邪�A���ȋ쓮���q���f��[1]�ƘA���̕�����[2]�̓o��ɂ��A�����w�҂̊S���W�߂�悤�ɂȂ����B���̌�AVicsek���f�����͂��߂Ƃ��鎩�ȋ쓮���q�n�̏W�c�^���̐����ɂ��ė������i�W����ƂƂ��ɁA�o�N�e���A��^�j�זE�Ƃ������~�N���Ȑ����n�A����ɉ��U���̂�R���C�h�Ȃǔ���p���������n�ł��̔t�_�C�i�~�N�X�����ׂ��Ă���[3]�B����A�����̌Q��^���ɂ��Ă͉f����GPS���M���O��p�����ϑ��ɂ��A���[�_�[�^�t�H�����[�W�ȂǁA��蕡�G�ȑ��ݍ�p�𐄒肷�邱�Ƃ��\�ƂȂ���[4,5]�B���̏ꍇ�A�̂̈ӎv����̌Q������ł̓`�d���l�b�g���[�N�Ƃ��ĕ\�����A���͂��邱�Ƃ��ł���B�{���\�ł́A���������W�c�^���E�W�c�I�ӎv����ɂ��Ă̌��������r���[����Ƃ��ɁA���\�҂��n���K���[�؍ݒ��ɍs�����C���^�[���V�b�v�̐��ʂɂ��ĕ���B
[1] T. Vicsek, A. Czirok, E. Ben-Jacob, I. Cohen, and O. Shochet, Phys. Rev. Lett. 75, 1226 (1995).
[2] J. Toner and Y. Tu, Phys. Rev. Lett. 75, 4326 (1995).
[3] S. Ramaswamy, Annu. Rev. Condens. Matter Phys. 1, 323 (2010).
[4] M. Nagy, Z. Akos, D. Biro, and T. Vicsek, Nature 464, 890 (2010).
[5] U. Lopez, J. Gautrais, I. D. Couzin, and G. Theraulaz, Interface Focus 2, 693 (2012).
�wPolyexciton stability in multi-valley semiconductor and optical trap for valley exciton�x
Professional development Consortium for Computational Materials Scientists (PCoMS) is the organization established to develop human resources of computational material science. I participated the internship program of PCoMS and stayed in prof. Varga's group in the Vanderbilt University for almost 1 month in this spring. In this stay. My activity was focused on settling the current research project and putting the next research issue into shape. Current project is the identification of polyexciton stability in multi-band semiconductor and verified up to triexciton bound states by numerical calculation. Next research issue is assessing the feasibility of optical trap for 2D exciton by utilizing optical Stark effect. I will report the details and contributions of the internship to my research.
�y2015�N�x�z
It is quite common to perform a supercell calculation for systems containing impurities, vacancies, lattice distortion, or long-range orders, where the translational symmetry is broken. The imperfections not only change the band structures of the original systems but also introduce a large amount of horizontal-like bands due to the band folding from the larger zones into the smaller supercell zones. How heavily the bands are folded to mess up the original band structures depends on how large the supercells are. Of course, the "mess up" also depends on how strong the degree of translational symmetry breaking is. However, the appearance of heavily folded bands is not the case in experimental observations. The measured spectral weight cannot reveal those heavily folded bands even by ideally switching on the small perturbations in their samples. Along this line, most supercell states should carry negligible spectral weight. For having a reasonable comparison with experiments and better visualization to understand symmetry breaking theoretically, I will talk about how to unfold the supercell bands into a larger Brillouin zone.[1, 2] The reference Brillouin zone could be chosen to be larger than the Brillouin zone of the primitive unit cell containing no imperfection. Several examples will be given in the talk. For example, by considering there exists only one lattice in systems like silicene instead of commonly treated two sub-lattices, the two atoms in the primitive unit cell can interfere with each other and cancel the spectral weight in some region in the reciprocal space. The choice of one-Si-atom Brillouin zone can demonstrate good agreement of spectral weight with the ARPES measurement in the case of silicene on ZrB2 thin film.[3]
[1] Wei Ku et al., Phys. Rev. Lett. 104, 216401 (2010).
[2] Chi-Cheng Lee et al., J. Phys.: Condens. Matter 25, 345501 (2013).
[3] Chi-Cheng Lee et al., Phys. Rev. B 90, 075422 (2014).
�wFirst-principles study of large spin-orbit coupling transition-metal compounds: electronic structure and new possibilities�x
Recently Sr2IrO4 has attracted considerable attention due to the intriguing interplay between spin and orbital degrees of freedom, and its manifestation in the material characteristics. Along this line I will present our recent progress on the iridium oxide and other related compounds based on 4d-/-5d- transition metals. The first example is Rh-doped iridate, Sr2Ir1-xRhxO4, for which the doping dependent metal-insulator transition (MIT) has been reported experimentally and the controversial discussion developed regarding the origin of this transition [1]. We tried to suggest a new picture for understanding the MIT. The second is the artificially-structured iridate superlattice, SrIrO3/SrTiO3. The electronic structure change caused by the interface and the strain was examined in detail and compared with the optical spectra [2]. Finally, I will introduce a fairly different family of compounds, GaT4X8 (T=Nb, Mo, Ta, and X=Se,Te). Interesting similarities with Sr2IrO4 suggests a new possibility for the novel ground states in this series of compounds [3].
[1] Chakara, et al., (submitted)
[2] K. -H. Kim, H. -S. Kim, and M. J. Han, J. Phys.: Condens. Matter 26, 185501 (2014)
[3] H. -S. Kim, J. Im, M. J. Han, H. Jin, Nature Comm. 5, 3988 (2014)
�wInteratomic potential of iron based on machine learning�x
Prediction of crystal structures for alloys and compounds with arbitrary composition of elements has not been widely performed yet, while current first-principles electronic structure calculations based on density functional theories (DFT) might have enough accuracy in predicting crystal structures in most cases. One of reasons for that is due to a large computational cost to explore vast configuration space in structural prediction by using molecular dynamics and Monte Carlo simulations based on DFT. In this talk, we report an ongoing research to develop an accurate interatomic potential of iron using a neural network (NN) method being a machine learning technique as a first step towards structural prediction of magnetic intermetallic compounds. Behler et al. proposed a way of developing accurate interatomic potentials using NN by letting NN learn a series of DFT calculations [1]. In addition to two- and three-body structural symmetry functions as structural fingerprint proposed by Behler, we further introduce four kinds of spin symmetry functions to develop interatomic potentials using NN which can treat crystal and spin structures on the same footing. The total energies of reference systems with an arbitrary spin structure, BCC, FCC, HCP, simple cubic, diamond, and their distored structures with ferro-, antiferro-, and non-magnetic, random spin structures, were carefully calculated by using a constraint scheme, which has been newly developed, based on non-collinear DFT [2], and all the calculated data were assembled to form a database as reference. By providing the database as a trainnig set, we trained NN with a feed-forward network structure of four layers using a back propagation, stochastic mini-batch decent, and residual minimization method. It is found that the mean difference between the reference DFT data and NN is 9.6 meV/atom, which suggests that a highly accurate interatomic potential can be developed for magnetic systems. We also discuss how the potential can be utilized to simulate crystal and spin dynamics on the same footing.
[1] J. Behler and M. Parrinello, Phys. Rev. Lett. 98, 146401.
[2] http://www.openmx-square.org/
�wLarge-scale ab initio simulations based on systematically improvable atomic basis�x
Atomic orbitals have many advantages as basis sets for ab initio electronic structure calculations. They have been popular in recently developed fist-principles simulation packages. However, the atomic basis sets must be constructed very carefully to ensure both good accuracy and transferability. Furthermore, the quality of the basis sets should be systematically improvable in an unbiased way. We have proposed a unique scheme to construct systematically improvable optimized atomic basis sets. This scheme has been implemented in our home made first-principles packages ABACUS. Our benchmark tests show that our atomic bases work very well for wide range of physical systems, including bulks, molecules, surfaces, defects, etc.
�y2014�N�x�z
�J�ڋ��������ɂ����鎥�C�ٕ����⎥�ǁE�X�L���~�I���ȂǃX�s���e�N�X�`���� ���_�I�\���Ƃ��̕����T���͉��p��ɂ߂ďd�v�Ȉʒu�Â��ɂ���B���̔w�i�̂��ƁA �m���R���j�A�����y�уX�s���O�����ݍ�p���l�������S�d�q�t���|�e���V���� ���`���⋭���ʔg�i�e�k�`�o�v�j�@���J�����A�J�ڋ��������₻�̕\�ʊE�ʂȂ� �����n�֓K�p�����݂Ă����B�u���ł́A�J�ڋ��������E�\�ʊE�ʂɂ����鎥�ǂ� �����X�p�C�����A���C�Q�Ȃǂ̃X�s���e�N�X�`���̍\�����萫�A����������Â��� ���C�I���ݍ�p�́i�������ݍ�p�́A�������C�ٕ����W���A�W�����V���X�L�[�E ��J���ݍ�p�́j�A�����̓d�E����Ɋւ��邱��܂ł̌v�Z����[1]�ɂ��ďЉ��B
[1] Nakamura, et. al., PRB 68, 180404R (2003); PRL 93, 057202 (2004); PRL 102, 187201 (2009), Hotta et.al, PRL 110, 267206 (2013), Phark et.al., Nat. Commu. 5, 5183 (2014).
�wTheory Seminar:Theoretical and Experimental Exploration of Two-Dimensional Silicon Structures �x
Although it is believed that two-dimensional honeycomb structures consisting of silicon atoms do not exist experimentally due to relative instability of its hybridized sp2 orbitals, a recent experiment clearly demonstrates that silicene, honeycomb structure of silicon atoms, can be fabricated on ZrB2 (0001) thin films [1]. Here, we report on detailed studies for geometrical and electronic structures of silicene on ZrB2 and a related two-dimensional structure by means of electronic structure calculations based on density functional theories (DFT), guided by a close collaboration with experiments performed by the Yamada-Takamura group of JAIST [1-6]. Theoretical chemical shift of Si-2p states [1] and band structure calculations [4] strongly support the formation of silicene having a planar-like structure. The stability of the planar-like structure over the regularly buckled structure can be understood by interaction between states of the silicene and surface states consisting of the d-orbital of the top Zr atoms [2]. We also propose a possible mechanism for the formation of the domain structure of silicene on ZrB2 [1,5]. It is inferred that the domain structure is induced by an instability of a phonon having a nearly zero frequency, and is formed in such a way that the k-points having the zero frequency can be removed from the first Brillouin zone. The mechanism is verified by performing large-scale total energy calculations. We further explore a possible structure of multi-layer silicene, and find that the MoS2 structure consisting of silicon atoms is stabilized with atoms in the inner layer having a sixfold coordination, which results in cigar-shaped nematic orbitals originating from the Si-sp2 orbitals [6]. [1] A. Fleurence, R. Friedlein, T. Ozaki, H. Kawai, Y. Wang, and Y. Yamada-Takamura, Phys. Rev. Lett. 108, 245501 (2012).
[2] C.-C. Lee, A. Fleurence, R. Friedlein, Y. Yamada-Takamura, and T. Ozaki, Phys. Rev. B 88, 165404 (2013).
[3] A. Fleurence, Y. Yoshida, C.-C. Lee, T. Ozaki, Y. Yamada-Takamura, and Y. Hasegawa, Appl. Phys. Lett. 104, 021605 (2014).
[4] C.-C. Lee, A. Fleurence, Y. Yamada-Takamura, T. Ozaki, and R. Friedlein, Phys. Rev. B 90, 075422 (2014).
[5] C.-C. Lee, A. Fleurence, R. Friedlein, Y. Yamada-Takamura, and T. Ozaki, submitted to Phys. Rev. Lett.; arXiv:1408.2588.
[6] F. Gimbert, C.-C. Lee, R. Friedlein, A. Fleurence, Y. Yamada-Takamura, and T. Ozaki, Phys. Rev. B, in press; arXiv:1401.0142.