

Research Highlights:
Optimized atomic orbitals for large-scale electronic structures
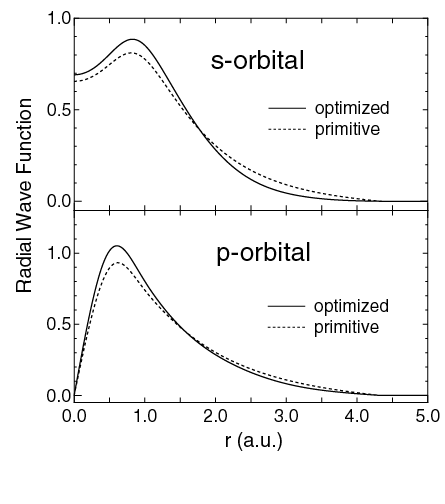
orbitals of a carbon atom in the diamond structure.
In order to extend the applicability of density functional theories (DFT) to realistic large systems, considerable efforts have been made for developing O(N) methods of the eigenvalue problem and for making efficient and accurate localized orbitals as a basis set being suitable for O(N) methods. Among these studies, one of important problems is how atomic orbitals as a basis set are constructed to maximize both the computational efficiency and accuracy. To address the issue, we have developed a simple and practical method for variationally optimizing numerical atomic orbitals based on the force theorem [1]. The derived equation provides the same procedure for the optimization of atomic orbitals as that for the geometry optimization. The optimized orbitals well reproduce convergent results calculated by a larger number of unoptimized orbitals. In addition, we demonstrate that the optimized orbitals significantly reduce the computational effort in the geometry optimization, while keeping a high degree of accuracy. Using the method we successfully constructed a set of optimized basis functions for biological molecules such as proteins, polysaccharides, and deoxyribonucleic acid [2]. We further performed the first systematic study for numerical atomic basis orbitals ranging from H to Kr, which could be used in large scale O(N) electronic structure calculations based on DFT. The comprehensive investigation of convergence properties with respect to our primitive basis orbitals provides a practical guideline in an optimum choice of basis sets for each element, which well balances the computational efficiency and accuracy [3]. Based on these methodological developments, we recently constructed a database of optimized basis functions as Database (2011) [4]. which is widely used for a variety of problems. The research was conducted in collaboration with NIMS.
- "Variationally optimized atomic orbitals for large-scale electronic structures", T. Ozaki, Phys. Rev. B 67, 155108 (2003).
- "Numerical atomic basis orbitals from H to Kr", T. Ozaki and H. Kino, Phys. Rev. B 69, 195113 (2004).
- "Variationally optimized basis orbitals for biological molecules", T. Ozaki and H. Kino, J. Chem. Phys. (2004).
- "Database (2011) of optimized VPS and PAO", T. Ozaki.