

Research Highlights:
大規模電子状態計算のためのO(N)クリロフ部分空間法
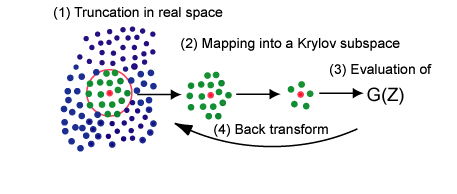
密度汎関数理論に基づく第一原理電子状態計算の計算コストは原子数Nの三乗に比例して増大することが知られており、 一般にO(N3)法と呼ばれています。 これはある参照系から比較し、10倍大きな系を計算する場合、その計算コストが1000倍になることを 意味しています。工学分野で扱われる系は、固液界面、粒界、表面修飾された金属クラスター等、非常に複雑な構造を 持っており、数千から数万原子程度の計算が必要となってきます。しかしながら、O(N3)法ではその計算コスト のため、その様な大規模系の取扱いは簡単ではありません。 その一方で、分子における官能基の化学的性質の大部分は局所的な原子配置によって決定されていることが知られています。 この化学的直感に基づき、おもに近距離からの情報を利用することで、局所的な電子状態を決定する効率的な近似計算手法を 開発することが可能です。その様な近似計算手法の計算コストは原子数Nに比例することからO(N)法と呼ばれています。
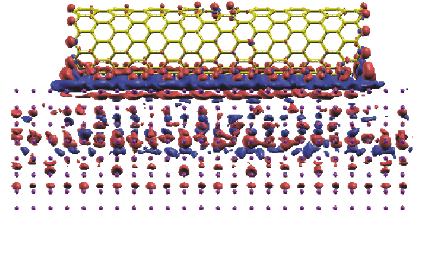
置かれた(10,0)ジクザグカーボンナノチューブ系の差電子密度