

Research Highlights:
大規模電子状態計算のための最適化数値局在基底関数
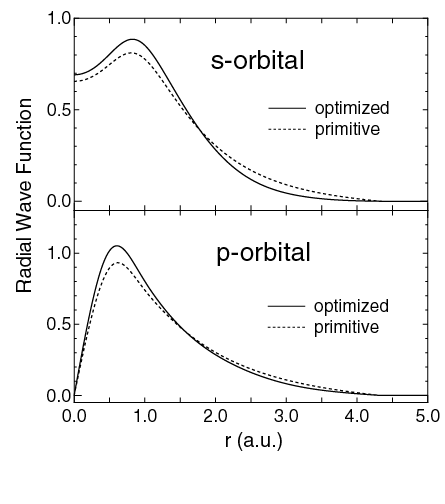
密度汎関数理論(DFT)の大規模系への適用を拡大するために、O(N) 固有値計算手法や、O(N)法に適した基底セットとして効率的かつ高精度な局在軌道の作成に多大な努力が払われてきました。これらの研究の中で、計算効率と計算精度の両者を最適化するために、基底セットとしての原子軌道をどのように構築するかは重要な問題の一つです。この問題を解決するために、我々は力の定理 [1]に基づいて、数値的な擬原子軌道を変分的に最適化するための実用的な方法を開発した。導出されたアルゴリズムは、構造最適化と同じ手順で数値局在軌道を最適化します。最適化された軌道は,より多くのプリミティブ軌道によって計算された収束結果をよく再現しています。さらに、最適化軌道を用いることで、高い精度を保持したまま構造最適化の計算量を大幅に削減できることを実証しました。この手法を用いて、タンパク質,多糖類,デオキシリボ核酸などの生体分子に最適化された基底関数のセットを構築することに成功しました [2]。さらに, DFTに基づく大規模なO(N)電子構造計算で使用できるHからKrまでの数値原子基底軌道の体系的な研究を実行しました [3]。プリミティブ基底関数に対する収束特性の包括的な調査から、適切な基底関数系の選択における実用的なガイドラインが得られました [3]。これらの方法論の発展に基づいて、私たちは最近、最適化された基底関数と擬ポテンシャルのデータベースを構築しました[4]。このデータベースは様々な問題に広く活用されています。本研究はNIMSと共同で実施されました。
- "Variationally optimized atomic orbitals for large-scale electronic structures", T. Ozaki, Phys. Rev. B 67, 155108 (2003).
- "Variationally optimized basis orbitals for biological molecules", T. Ozaki and H. Kino, J. Chem. Phys. (2004).
- "Numerical atomic basis orbitals from H to Kr", T. Ozaki and H. Kino, Phys. Rev. B 69, 195113 (2004).
- https://t-ozaki.issp.u-tokyo.ac.jp/vps_pao2019/, T. Ozaki.